Rice plants and growth conditions
We used the rice ecotype NPB or DJ as the wild-type control. The coi1-13 mutant was identified by quantitative PCR with reverse transcription (RT–qPCR) and phenotypic observation, consistent with previous reports59. The jamyb mutant was identified by sequencing, as previously described17. The ninja1 (modd-2) mutant was identified by genotyping using gene-specific and T-DNA-specific primers as described previously46. All of the rice plants were cultivated in a greenhouse at 28–32 °C and at a relative humidity of 60 ± 5%.
N. benthamiana growth conditions
N. benthamiana plants were cultivated in soil within a greenhouse under a 16 h–8 h light–dark photoperiod at a constant temperature of 24 °C. For transient expression assays, the upper three leaves of 1-month-old plants were used.
Vector construction and rice transformation
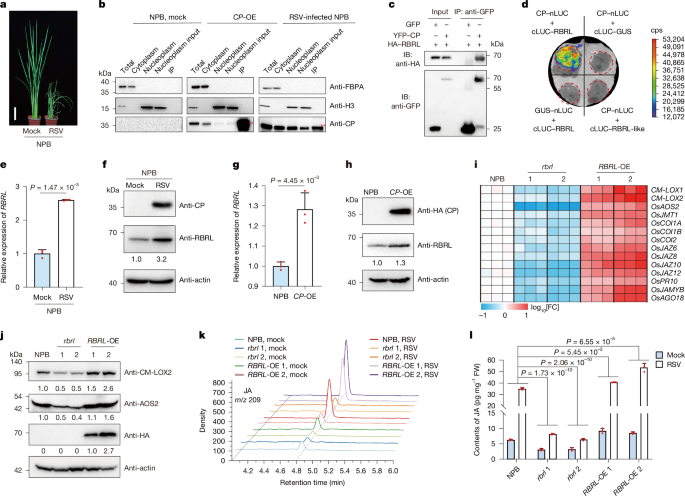
The coding sequences of OsRBRL, RSV CP, OsNINJA2 and OsNINJA3 were initially amplified by RT–PCR. Subsequently, the genes were individually cloned and inserted into the pCambia2300-Actin1-OCS and pCambia2300-Actin1-3×MYC vectors, resulting in the generation of the Actin1::HA-OsRBRL, Actin1::HA-CP, Actin1::3×MYC-OsNINJA2 and Actin1::3×MYC-OsNINJA3 constructs. The rbrl-, ninja2-, ninja3- and ninja1/2/3-knockout lines were generated using CRISPR–Cas9 according to established protocols60. These constructs were subsequently introduced into wild-type NPB plants or rbrl 2 mutant plants through Agrobacterium tumefaciens-mediated transformation using a process facilitated by BioRun and BIOGLE GeneTech. The rice lines overexpressing OsRBRL, RSV CP and OsNINJA3 were verified by immunoblotting. The genome-edited mutants were confirmed by sequencing and analysed using SnapGene (https://www.snapgene.com). For transient expression assays, genes in recombinant binary vectors were initially amplified by RT–PCR and subsequently cloned and inserted into the corresponding entry vectors pEASY-Blunt-Simple and pENTR/D-TOPO. These inserts were subsequently cut and either ligated or recombined into destination vectors. Detailed primer information is provided in Supplementary Table 5.
Immunoblotting and quantification analysis
Plant samples were homogenized in liquid nitrogen, and total proteins were extracted from equal weights of ground powder using the same volume of 2× Laemmli buffer (4% (w/v) SDS, 20% (v/v) glycerol, 10% (v/v) 2-mercaptoethanol, 0.004% (w/v) bromophenol blue and 0.125 M Tris-HCl, pH 6.8) and subsequently boiled at 95 °C for 10 min. The supernatants were collected by centrifugation at 12,000 rpm for 5 min and separated by SDS–PAGE. The PageRuler Prestained Protein Ladder (Thermo Fisher Scientific) and Prestained Protein Ladder (Meilun Biotech) were used as molecular mass standards. The proteins were subsequently transferred to nitrocellulose membranes and detected with antibodies against CM-LOX2 (1:2,000)17, AOS2 (1:2,000)17, RSV CP (1:5,000)61, RDV P2 (1:5,000)62, AGO18 (1:500)18, AO (1:500)19, RBRL (1:500), NINJA3 (1:500), MYC (ABclonal, 1:2,000), HA (ABclonal, 1:2,000), GFP/YFP (ABclonal, 1:2,000), GST (ABclonal, 1:2,000), MBP (ABclonal, 1:2,000), Flag (TransGen Biotech, 1:2,000), S (EarthOx, 1:2,000), FBPA (Beijing Protein Innovation, 1:2,000), histone H3 (Abcam, 1:2,000), cLUC (Sigma-Aldrich, 1:1,000) and actin (CWBIO, 1:10,000). Actin was used as the loading control or sample processing control. Images from immunoblotting were collected using the Molecular lmager ChemiDoc XRS+ (Bio-Rad) system. The corresponding band intensities were quantified using ImageJ (https://imagej.net/ij/). The band intensities for a particular protein were normalized to those for actin or Rubisco. Relative values were calculated by comparison with the first band on the left in each figure. Uncropped immunoblotting images are provided in Supplementary Fig. 1.
Nuclear‒cytoplasmic fractionation followed by an immunoprecipitation assay
To precisely screen for rice proteins that interact with RSV CP in the nucleus, we performed nuclear–cytoplasmic fractionation followed by IP–MS on RSV-infected NPB and CP-OE rice, with mock NPB rice used as a control. First, we conducted subcellular fractionation on the rice materials with minor modifications as described previously63. The rice materials were harvested and ground into powder in liquid nitrogen at 4 weeks after RSV infection. The powder (approximately 1 g) was resuspended in Honda buffer (0.4 M sucrose, 2.5% Ficoll, 5% dextran T40, 25 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 0.5% Triton X-100, 0.5 mM PMSF, 10 mM β-mercaptoethanol, RNase inhibitor and Roche protease inhibitor cocktail) at a ratio of 2 ml g−1. The homogenate was filtered twice through a double-layered Miracloth, and the supernatant was subsequently centrifuged at 1,500g for 5 min at 4 °C. The supernatant was further centrifuged at 10,000g for 10 min at 4 °C to obtain the cytoplasmic fraction. The pellet containing the nuclear proteins was resuspended in three volumes of nuclear extraction buffer (500 mM KCl, 20 mM Tris-HCl (pH 8.0), 0.5% Triton X-100, 25% glycerol, 1.5 mM MgCl2, 0.5 mM EDTA, 1 mM dithiothreitol (DTT), cocktail and 1 mM PMSF) and incubated for 30 min at 4 °C. Insoluble nuclear residues were then sedimented at 10,000g at 4 °C for 10 min, and the supernatant was collected for the IP assay. The nuclear extract was diluted with four volumes of dilution buffer (20 mM Tris-HCl (pH 8.0), 500 mM KCl, 0.5% Triton X-100, 1 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, cocktail and 1 mM PMSF). The mixture was precleared with protein G for 30 min, and 1:100 anti-CP antibody was then added and incubated for 2 h, followed by the addition of 1:200 protein G and further incubation for 2 h. The samples were washed three times with washing buffer (20 mM Tris-HCl (pH 8.0), 500 mM KCl, 0.5% Triton X-100, 5% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, cocktail and 1 mM PMSF). Finally, the bead-bound proteins were eluted in 2× Laemmli buffer and subjected to SDS–PAGE. For LC–MS/MS analysis, the gel was subjected to silver staining to identify the candidate CP-interacting proteins.
IP for LC‒MS/MS assay
IP–LC–MS/MS assays were performed as previously described with some modifications64,65. To detect proteins that interact with RBRL, we performed IP-LC–MS/MS assays with transgenic plants that overexpressed HA-tagged versions of RBRL. In brief, rice samples were homogenized in liquid nitrogen. Total proteins were extracted from 200 mg of ground powder using IP buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 4 mM MgCl2, 0.5% (v/v) NP-40, 5 mM DTT, 1 mM phenylmethylsulfonyl fluoride, 50 μM MG132 and 1× protease inhibitor cocktail) and then incubated for 30 min at 4 °C with gentle rotation. Next, the samples were centrifuged at 12,000 rpm and 4 °C three times for 5 min each; the supernatants were collected after each centrifugation step. The resulting supernatant was transferred to a new tube containing 7.5 μl of HA-magnetic agarose (MBL) and incubated at 4 °C for 2.5 h with gentle rotation. The samples were then washed three times with wash buffer A (50 mM Tris-HCl (pH 7.4), 150 mM NaCl and 4 mM MgCl2). Finally, the bead-bound proteins were eluted in 2× Laemmli buffer and subjected to SDS–PAGE. For LC–MS/MS analysis, the gel was subjected to silver staining to identify the candidate RBRL-interacting proteins.
LC‒MS/MS assay
First, the gel was cut into small pieces and placed into tubes for digestion. A total of 400 μl of decolourization solution was added to each tube and shaken until completely decolorized, after which the liquid was discarded. Next, 400 μl of acetonitrile was added, and the mixture was shaken until the gel pieces turned white; the liquid was then discarded. Next, 200 μl of 10 mM DTT and 25 mM NH4HCO3 were added, and the mixture was incubated at 56 °C for 1 h. The mixture was cooled to room temperature, the liquid was discarded, 200 μl of 55 mM IAM and 25 mM NH4HCO3 were added, and the mixture was incubated in the dark for 45 min. After the liquid in the tubes was discarded, the gels were washed twice with 25 mM NH4HCO3. Next, 400 μl of acetonitrile was added, and the mixture was shaken until the gel pieces turned white. The liquid was discarded, and the gel pieces were crushed with a pipette tip. For each tube, trypsin was added to 25 mM NH4HCO3 buffer at a trypsin ratio of 1:50 (w/w), and the mixture was subsequently incubated at 37 °C overnight. Then, 200 μl of acetonitrile containing 0.1% formic acid was added to each tube, the tubes were shaken for 10 min and the supernatant was then transferred to a clean tube. Next, 30 μl of 0.1% formic acid was added to the gel, which was shaken for 10 min, then 200 μl of acetonitrile containing 0.1% formic acid was added and the mixture was shaken for another 10 min. The supernatant was collected and dried using a vacuum centrifuge. Peptide samples were dissolved in 0.1% formic acid solution to a concentration of approximately 0.1 μg μl−1. The mixture was centrifuged at 16,700g for 12 min, and the supernatant was transferred to a MS injection vial to conduct the LC–MS/MS assay. LC–MS/MS analysis was performed using the EASY-nLC 1200 liquid chromatography system and a C18 column coupled with a Thermo Fusion Lumos mass spectrometer. The mobile phase for liquid chromatography consisted of 0.1% formic acid (phase A) and 80% acetonitrile/0.1% formic acid (phase B). The flow rate was set at 280 nl min−1. The LC–MS/MS results were processed using Proteome Discoverer 2.2 software and the rice database to acquire the CP- and RBRL-interacting proteins.
Transient expression in N. benthamiana leaves
Agrobacterium-mediated transient expression assays were performed as previously described17 with minor modifications. The recombinant binary vectors were subsequently transformed into A. tumefaciens strain GV3101 using the freeze–thaw method. Suspensions of Agrobacterium cultures were adjusted to an optical density at 600 nm (OD600) of 1.0 and used to infiltrate leaves of N. benthamiana plants at the four- to five-leaf stage through a 1-ml syringe without a needle. Leaf tissues were harvested at 2 days after agroinfiltration.
LCI assay
LCI assays were also conducted as previously described17. The coding sequences of OsRBRL, RSV CP, four OsNINJA genes and OsRBRL-like, and the RBR NTD domain, RBR RBR domain, RBR Ari domain, NINJA3 EAR domain, NINJA3 NINJA-B domain, NINJA3 ZBD NTE domain and NINJA3 ZBD core domain and all OsJAZ genes were separately inserted into the pCambia1300-nLUC or pCambia1300-cLUC vectors. A list of the primers used is provided in Supplementary Table 5. All of the constructs were subsequently transformed into A. tumefaciens strain GV3101 using the freeze–thaw method. Four combinations of A. tumefaciens suspensions were mixed, adjusted to a final concentration with an OD600 of 1.0 and coinfiltrated into four different regions on the same N. benthamiana leaf. At 2 days after agroinfiltration, cells were infiltrated with 200 μM luciferin (Promega), and luciferase activity was detected using a low-light cooled charge-coupled device imaging apparatus (NightOWL II LB983 with IndiGO software v.2.0.4.0). At least three biological replicates were performed.
Co-IP assay
35S::YFP-CP was constructed by inserting the coding sequence of RSV CP into the pEarleyGate104 vector and was subsequently used to express YFP–CP. 35S::HA-RBRL was constructed by fusing the coding sequence of OsRBRL into the pCambia2300 vector and was subsequently used to express HA–RBRL. pCambia1301-35S-GFP was used to express GFP. All of the constructs were subsequently transformed into A. tumefaciens strain GV3101 using the freeze–thaw method. The indicated combinations of A. tumefaciens suspensions were mixed to a final concentration with an OD600 of 1.0. The infiltration procedures were performed as described above (see the ‘Transient expression in N. benthamiana leaves’ section). Leaves were harvested at 2 days after agroinfiltration, and total proteins were extracted with native extraction buffer (50 mM Tris-MES (pH 8.0), 500 mM sucrose, 1 mM MgCl2, 10 mM EDTA, freshly added 5 mM DTT and 1× protease inhibitor cocktail)66. The mixture was incubated at 4 °C for 30 min with gentle rotation and then centrifuged at 12,000 rpm and 4 °C three times for 10 min each; the supernatants were collected after each centrifugation step. Cleared extracts were immunoprecipitated using GFP-tagged or HA-tagged Nanoselector Agarose (HUABIO) and incubated for 1 h at 4 °C with gentle rotation. The samples were then washed three times with washing buffer B (10 mM Tris-HCl (pH 7.5), 150 mM NaCl and 0.5 mM EDTA). Finally, the bead-bound proteins were eluted in 2× Laemmli buffer. The eluted proteins were boiled for 10 min, centrifuged, separated by SDS–PAGE and detected with antibodies against HA (ABclonal) and GFP (ABclonal).
Protein expression and purification
The coding sequences of RSV CP, RDV P2, RDV P21–786, RDV P9, OsRBRL and four OsNINJA genes were inserted into the pHM4 (MBP tag, modified from pMAL-c2X Vector, New England Biolabs), pGEX4T1 (GST tag, GE Healthcare), pACYCDuet42 or pCDFDuet42 vector and expressed as tag fusion proteins (MBP–RBRL, GST–CP, GST–NINJA1, GST–NINJA2, GST–NINJA3, GST–NINJA4, RBRL–MYC, MBP–P2–HA, MBP–P9–HA, MBP–P21–786 and MBP–NINJA3–HA) in E. coli strain Transetta or BL21 (DE3; TransGen Biotech). The fusion proteins were purified using glutathione Sepharose 4B beads (GE Healthcare) or amylose resin (New England Biolabs) or detected by immunoblotting as previously described42.
Pull-down assay
Equimolar amounts of MBP–RBRL on MBP tag Nanoselector Agarose (HUABIO) were separately incubated with equal amounts of GST, GST–CP, GST–NINJA1, GST–NINJA2, GST–NINJA3 or GST–NINJA4 in pull-down buffer (20 mM Tris-HCl (pH 7.5), 200 mM NaCl) at 4 °C for 1 h with gentle rotation. The beads were then washed (five times for 5 min each) with pull-down buffer (containing 2% (v/v) Triton X-100). The bound proteins were boiled in 2× Laemmli buffer, separated by SDS–PAGE, and detected with antibodies against MBP (ABclonal) and GST (ABclonal).
Virus inoculation
The virus inoculation procedures were performed as previously described17,67. In brief, 2-week-old rice plants were inoculated with two viruliferous (containing RSV or RDV) or virus-free (mock) insects. Then, 2 days after inoculation, the insects were removed, and the rice plants were returned to the greenhouse as described above (see the ‘Rice plants and growth conditions’ section). The inoculated plants were monitored weekly for the appearance of viral symptoms. The numbers of rice plants of each line with various disease symptom grades were recorded at 4 w.p.i. (Supplementary Table 3). Photographs of plants with representative symptoms were acquired at 2 or 4 w.p.i.; whole shoots were harvested for RT–qPCR and immunoblotting assays at 4 w.p.i.
RNA extraction and RT‒qPCR analysis
Rice samples were homogenized in liquid nitrogen, and total RNA was extracted from 100 mg of ground powder using TRIzol Reagent (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. Total RNA was treated with RQ1 RNase-free DNase (Promega) to remove traces of contaminating genomic DNA. The RNA was subsequently reverse-transcribed using M-MLV reverse transcriptase (Promega), oligo(dT)18 primer and recombinant RNasin ribonuclease inhibitor (Promega) according to the manufacturer’s instructions. The resulting cDNA was used as the template for RT–PCR and RT–qPCR. RT–qPCR was performed using the SYBR Green Real-Time PCR Master Mix (Mei5 Biotech) according to the manufacturer’s instructions using the Bio-Rad CFX96 system with CFX Maestro 1.1 software. The level of OsEF-1a expression was detected in parallel and used as the internal control. A list of the primers used is provided in Supplementary Table 5.
Phylogenetic analysis
The amino acid sequences of RBRL, other RBR family proteins, and NINJA family proteins were obtained from UniProt (https://www.uniprot.org/). An unrooted, neighbour-joining tree was constructed using Molecular Evolutionary Genetic Analysis 11 (MEGA11).
Generation of rice RBRL and NINJA3 antibodies
Rabbit polyclonal antibodies were generated by HUABIO or ABclonal. The synthetic peptides RBRL (DLHLRLPDDRPADC) and NINJA3 (STGKPLNGTVTQQS) were used to obtain rabbit polyclonal antibodies to RBRL and NINJA3, respectively. The antisera were affinity-purified and used for immunoblotting.
Sample preparation and JA quantitative assay
Hormone extractions and measurements of the JA concentration were performed as previously described68. In brief, rice shoots from 2-week-old NPB, rbrl and RBRL-OE plants were separately collected and ground into fine powder. JA was extracted from 110 mg of ground powder using 400 μl of 10% methanol containing 1% acetic acid and then purified with a 0.22-μm nylon filter. The eluate was analysed by ultra-high-performance liquid chromatography–triple quadrupole MS (UPLC–MS/MS) on a mass spectrometer (UPLC 1290-MS/MS 6495).
In vivo and semi-in vivo protein degradation
In vivo and semi-in vivo protein degradation experiments were conducted as previously described66. For in vivo protein degradation experiments, agrobacterial strains containing 35S::HA-RBRL, 35S::HA-RBRL-like, 35S::YFP-NINJA3, 35S::YFP-NINJA4, 35S::FLAG-GUSA (internal control) or 35S::MYC-CP were coinfiltrated at the indicated ratios. The infiltration procedures were performed as described above (see the ‘Transient expression in N. benthamiana leaves’ section). Then, 2 days after infiltration, the samples were collected for analysis. For semi-in vivo protein degradation experiments, agrobacterial strains containing 35S::HA-RBRL, 35S::YFP-NINJA3, 35S::YFP-NINJA4, 35S::GFP, 35S::YFP-CP or pCambia2300 (empty vector) were infiltrated separately. The infiltration procedures were performed as described above (see the ‘Transient expression in N. benthamiana leaves’ section). Then, 2 days after infiltration, the samples were collected separately and extracted using native extraction buffer (containing 10 μM ATP). An equal volume of the corresponding extracts was mixed together. A final concentration of 50 μM MG132 was added to the corresponding mixture. The mixtures were incubated at 4 °C with gentle rotation. The samples were removed at various timepoints, and the reaction was terminated by the addition of 2× Laemmli buffer and boiling for 5 min; finally, the samples were subjected to immunoblotting analysis.
Y2H assay
The coding sequences of 14 OsJAZ genes, 4 OsNINJA genes, 12 RDV proteins and OsRBRL were separately cloned and inserted into the pDEST-GBKT7 or pDEST-GADT7 vector. A list of the primers used is provided in Supplementary Table 5. Yeast transformation was performed as described by the vector manufacturer (Clontech, Mountain View). Different combinations of constructs were cotransformed into yeast AH109 cells. All yeast transformants were subsequently grown on SD/−Leu/−Trp and subsequently transferred to SD/−Leu/−Trp/−His/−Ade medium for interaction tests.
Transient expression in rice protoplasts
Transient expression in rice protoplasts was conducted as previously described, with some modifications17,65,69,70. In brief, the corresponding plasmids were cotransformed into protoplasts using polyethylene glycol (PEG). Rice protoplasts were isolated from the leaf sheaths of 10–14-day-old wild-type (NPB) or rbrl 2 plants. The leaf sheaths were initially cut into 0.5-mm pieces with sharp razor blades and then submerged in enzyme solution (0.4 M d-mannitol, 20 mM MES-KOH (pH 5.7), 20 mM KCl, 1.5% cellulase R10 (w/v), 0.7% macerozyme R10 (w/v), 0.1% bovine serum albumin and 10 mM CaCl2) for 7 h with shaking (40 rpm) at 28 °C in the dark. Each sample was filtered through a 40-μm nylon mesh filter. After removal of the enzyme solution, the tissues were suspended in W5 solution (154 mM NaCl, 125 mM CaCl2, 5 mM KCl and 2 mM MES (pH 5.7)) and subsequently filtered through another 40-μm nylon mesh filter. The flow-through samples from the above-mentioned filtrations were mixed and then centrifuged for 3 min at 900 rpm to pellet the protoplasts. The protoplasts were resuspended in W5 solution and incubated on ice for 30 min. The protoplasts were then centrifuged at 900 rpm for 3 min and resuspended at 3 × 105 cells per ml in MMG solution (0.4 M d-mannitol, 15 mM MgCl2 and 4 mM MES-KOH (pH 5.7)) for PEG-mediated transformation. In the transformation, 110 μl of freshly prepared PEG-CaCl2 solution (0.2 M d-mannitol, 100 mM CaCl2 and 40% (v/v) PEG 4000) and 10 μg (10 μl) of plasmid were gently mixed with 100 μl of protoplasts and then incubated for 15 min at room temperature in the dark. After transformation, the cells were washed with 10 volumes of W5 and then resuspended in W1 solution (0.5 M d-mannitol, 4 mM MES-KOH (pH 5.7) and 20 mM KCl) overnight at 28 °C in the dark. The GFP signal was quantified and normalized to the chlorophyll fluorescence, as previously reported71. The observation and quantification of each cell were performed under the same set of confocal parameters and at the same scale using LSM710 (Zeiss).
Dual-luciferase reporter system
The dual-luciferase reporter system was established as previously described17. In brief, the coding sequences of OsRBRL and OsRBRL-like were cloned and inserted into the pCambia2300-35S vector. A list of the primers used is provided in Supplementary Table 5. When conducting the assay in tobacco leaves, these two constructs were separately transformed into A. tumefaciens strain GV3101 using the freeze–thaw method. The indicated combinations of A. tumefaciens suspensions were mixed and adjusted to a final concentration with an OD600 of 1.0. The infiltration procedures were performed as described above (see the ‘Transient expression in N. benthamiana leaves’ section). The expression levels of firefly and Renilla luciferases in N. benthamiana leaves were measured using the GLO-MAX 20/20 luminometer (Promega) at 2 days after agroinfiltration. When conducting the assay in rice protoplasts, these two constructs were co-transformed into rice protoplasts along with 35S::myc-JAZ6, 35S::GFP, 35S::YFP-JAMYB, pCambia2300 (empty vector) and 35S::AGO18pro:LUC at a fixed ratio. The expression levels of firefly and Renilla luciferases in rice protoplasts were measured using the GLO-MAX 20/20 luminometer (Promega) at 12 h after transformation. The ratio of firefly luciferase to Renilla luciferase (LUC/REN) was calculated to determine the final transcriptional activity.
Virus isolation and rice protoplast infection
RSV was isolated and purified from rice plants infected with RSV, with some modifications compared to previous studies10. Approximately 10 g of leaves that had been stored at −80 °C were blended in 40 ml of phosphate buffer (0.1 M, pH 7.5) containing 0.1% 2-mercaptoethanol, 1% Triton X-100 and 0.01 M EDTA. The homogenate was then filtered through two layers of Miracloth (Millipore), mixed with 20% (v/v) chloroform and stirred for 15 min at room temperature. After centrifugation at 4 °C, 5,000g for 20 min for clarification, the supernatant was adjusted to 6% PEG 6,000 and 0.1 M NaCl in an ice bath for 4 h, and then rotated at 4 °C overnight. The resulting precipitate was collected by centrifugation at 4 °C, 5,000g for 20 min and the pellets were dissolved in 0.01 M phosphate buffer at pH 7.5. After centrifugation at 4 °C, 5,000g for 10 min, the supernatant was further centrifuged at 4 °C, 100,000g for 2 h. The resulting pellets were dissolved in 0.01 M phosphate buffer at pH 7.5. Carefully laid the resuspended pellet solution on top of 20% glycerol cushion. The virus particles were pelleted by centrifugation at 4 °C, 100,000g for 2 h. After centrifugation, discarded the supernatant and dissolved the pellets in 0.01 M phosphate buffer at pH 7.5. The purified virus particles were characterized by 10% SDS–PAGE, and the virus concentration was determined using the Bradford assay with BSA as a standard. The purified particles were then stored at −80 °C. When conducting rice protoplast infection, 1 μg RSV particles, 10 μg corresponding plasmid and 110 μl freshly prepared PEG-CaCl2 solution were gently mixed with 100 μl of protoplasts and then incubated for 15 min at room temperature in the dark. After transfection, the cells were washed with 10 volumes of W5 and then resuspended in W1 solution overnight at 28 °C in the dark. The infected protoplasts were collected at 18 h after infection and the RSV RNAs and RSV CP accumulation were analysed using RT–qPCR and immunoblotting.
Protein expression and ubiquitination assay in E. coli
The protein expression and ubiquitination assays conducted in the E. coli strain BL21 (DE3) were performed according to previous studies42. In brief, different combinations of the indicated expression vectors were transformed into the E. coli strain BL21 (DE3), and the strains were cultured in Luria–Bertani liquid medium at 37 °C. Then, 0.5 mM IPTG was added to the medium when the absorbance at 600 nm reached 0.4–0.6 to induce the expression of the target proteins. The bacteria were further cultured at 28 °C for 10–12 h and then stored at 4 °C overnight. The crude proteins were extracted and analysed by western blotting with the corresponding antibodies.
MST assay
The MST assays were conducted as previously described17. To assess the binding affinity between OsRBRL and RSV CP or RDV P2, we first labelled the GST–CP and MBP–P21–786 protein with the red fluorescent dye NHS according to the instructions of the Monolith Series Protein Labeling Kit RED-NHS 2nd Generation (NanoTemper Technologies). The NHS-labelled GST–CP and MBP–P21–786 protein was gradually diluted until its fluorescence intensity ranged between 800 and 1,000 under 20% LED power. The initial concentration of the MBP–OsRBRL protein was 258 µM. It was subjected to 12 rounds of 1:1 serial dilution to a final concentration of 0.126 µM. After brief incubation in the dark, the samples were loaded into MST standard capillaries. The measurements were performed with 20% MST power on a Microscale Thermophoresis Monolith NT.115 instrument (NanoTemper Technologies). The assays were repeated at least twice for each affinity measurement. Data analyses were performed using the MO.Affinity analysis software provided by the manufacturer.
Transcriptome sequencing
The aboveground parts of the mock- and RSV-infected rice plants were collected at 4 w.p.i. The plant material was thoroughly ground in liquid nitrogen and approximately 0.1 g of the sample was used for RNA extraction (see the ‘RNA extraction and RT–qPCR analysis’ section). Library construction and paired-end RNA-seq were performed by Biomarker Technologies. FastQC software was used to assess the quality of the raw sequencing reads. After adapters and low-quality reads were removed, the clean reads were mapped to the rice genome (MSU Rice Genome Annotation Project Database v.7.0, https://rice.uga.edu/download_osa1r7.shtml) using TopHat. Gene expression levels were calculated as fragments per kilobase per million reads. The multiple-testing-adjusted P value (false-discovery rate < 0.01) with an absolute fold change ≥ 2 was used to determine whether individual genes were significantly differentially expressed.
RBRL nuclear exclusion experiments
We conducted an RBRL nuclear-export experiment based on those performed in previous studies72. To prevent RBRL from localizing within the nucleus, a NES corresponding to amino acids 371 to 387 of Arabidopsis thaliana RTL2 (KKAESSSAYHMIRALRK) was introduced into the vector Ubi::RBRL-GFP. After sequencing verification, the modified construct was transformed into rice protoplasts with RSV particles (see the ‘Transient expression in rice protoplasts’ and ‘Virus isolation and rice protoplast infection’ sections).
Proteasome assembly assay
The proteasome assembly assays were conducted as previously described73. The aboveground parts of the mock- and RSV-infected NPB plants were collected at 4 w.p.i. The total protein extracted with buffer F (50 mM Tris-HCl (pH 7.5), 25 mM NaCl, 2 mM MgCl2, 1 mM EDTA, 2 mM DTT, 5 mM ATP and 5% glycerol) was used for native PAGE, followed by standard western blotting. Proteasome extracted from one-month-old A. thaliana Col-0 ecotype were used as a positive control and the marker. The anti-PAG1 antibody (1:1,000)73 was used to detect the assembly of proteasome.
Histochemical staining of H2O2 and O2·−
Hydrogen peroxide (H2O2) and superoxide (O2·−) in rice leaves were detected using the 3,3′-diaminobenzidine (DAB, Sigma-Aldrich) and nitro blue tetrazolium (NBT, Sigma-Aldrich) staining methods, with minor modifications based on previously described protocols19. In brief, rice leaves approximately 1 cm in length were immersed in either 10 mM Tris-HCl buffer (pH 6.5) containing DAB (1 mg ml−1) for H2O2 detection or 50 mM sodium phosphate buffer (pH 7.0) containing NBT (0.05%) for O2·− detection. The samples were incubated in the dark at 37 °C for 16 h. After incubation, the leaves were bleached with a solution of ethanol and acetic acid (3:1) at 70 °C for 60 min to remove chlorophyll. Finally, the leaves were washed 4–5 times with 75% ethanol until clear and photographed through a stereomicroscope under uniform lighting conditions.
H2O2 determination in rice seedlings
To quantify H2O2 levels in rice seedlings, a standardized protocol was used. Fresh rice leaves (100 mg) were weighed and ground into a fine powder using liquid nitrogen. The powder was mixed with 1 ml of 50 mM sodium phosphate buffer (pH 7.4) and incubated on ice for 30 min to facilitate H2O2 extraction. After incubation, the mixture was centrifuged at 13,000 rpm for 10 min at 4 °C and the supernatant was carefully transferred to a new tube. The H2O2 concentration was then determined using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen, A22188) according to the manufacturer’s instructions. The absorbance was measured at approximately 560 nm using a BioTek Microplate Reader (BioTek Cytation5), and the H2O2 content was calculated by comparing the sample absorbance to a standard curve of known H2O2 concentrations.
Quantification and statistical analysis
The data from the JA quantitative assays were analysed using two-way ANOVA. The data from the RT–qPCR analysis, dual-luciferase reporter system and protein degradation assay were analysed using Student’s t-tests or one-way ANOVA with Tukey’s multiple-comparison test. The above statistical analyses were performed with GraphPad Prism (v.7.0). All descriptive statistics of the JA quantitative analysis, RT–qPCR analysis, dual-luciferase reporter system and RSV disease symptom classification are shown as the mean ± s.d. The number (n) of biological replicates is indicated in each legend. For immunoblotting quantification, the band intensities were quantified using ImageJ. No statistical method was used to predetermine sample size. The sample sizes were determined from experimental trials and previous publications on similar experiments. Blinding and randomization were used. For example, the virus-infection assay, different rice lines were numbered; the investigator was blinded to the group allocation during the experiments including the inoculation of viruliferous small brown planthopper, the infection rates statistics, and virus RNAs RT–qPCR analysis. All samples were arranged randomly into experimental groups. Plants for experiments were grown side by side to minimize unexpected environmental variations during growth.
Statistics and reproducibility
Statistical analyses in Fig. 2g and Extended Data Figs. 3d,e and 4d were performed using one-way ANOVA with Tukey’s multiple-comparison tests, and different letters represent significantly different groups at the P < 0.05 level. Owing to the large number of P values obtained from pairwise comparisons, we list the exact P values in the corresponding source data as part of the Supplementary Information.
Material availability
All materials needed to replicate the work are available.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.


