Experimental model and study participant details
The sex, age and ethnicity of participants and their clinical manifestation details during acute MIS-C are reported in Extended Data Table 1 and in Supplementary Data 9–11 and 15, and for controls in Supplementary Data 8. Information on the gender of the patients with MIS-C and controls was not collected. The patients and controls were recruited in Berlin (Germany), Lyon (France), Ankara (Turkey), Santiago (Chile) and Napoli (Italy). This study was approved by the local institutional review boards (IRBs) of the Charité (Pa-COVID-19 and EA2/178/22) (Berlin, Germany), the Comité de Protection des Personnes Sud Méditerranée I (Marseille, France) (ID-RCB: 2020-A01102-37) for the French patients and by the ethical committee of Hospices civils de Lyon (Lyon University Hospitals, France) no. 23_5231 for French controls, by the Hacettepe University Ethical Committee (2021/09-45, Ankara, Turkey) for patients with MIS-C from Turkey, by Mass General Brigham IRB 2020P000955 for the patients and controls from Boston, and by the Ethics Committee Federico II in Naples as a collaborative study protocol with the NIH in 2020 (158/2020); and by the ethical committee of Clínica Alemana Universidad del Desarrollo (IRB ID 202098) for the samples from Chile. Written and informed consent was provided by legal representatives of the patients who participated in this study.
Human participants
Thirty-nine patients were recruited from the Charité Universitätsmedizin Berlin Department of Pediatric Respiratory Medicine, Immunology and Critical Care Medicine, 49 patients were recruited in Lyon and 20 patients were recruited in Ankara, Turkey, 2 patients in Naples, Italy and 14 patients in Santiago, Chile. MIS-C was diagnosed in all patients by a consultant paediatric rheumatologist according to the definitions from World Health Organization60, Centers for Disease Control and Prevention61 and Royal College of Paediatrics and Child Health62. Blood samples were taken during the acute phase of MIS-C and during follow-up visits in the outpatient clinic. Paediatric controls included in this study had a positive SARS-CoV-2 PCR result six weeks before inclusion. Additionally, samples from 36 healthy children, 57 children with asymptomatic or mild SARS-CoV-2 infection and 39 children with a moderate and 2 with a severe infection with SARS-CoV-2 during acute and follow-up and 11 children 6 wpi with SARS-CoV-2 were used as controls. Disease severity was defined according to the WHO clinical management guideline60.
Isolation of PBMCs and serum
Peripheral blood was drawn into EDTA collection tubes and SST tubes as previously described4. PBMC were isolated from peripheral blood by Ficoll-Paque PLUS (Cytiva) density gradient centrifugation at room temperature. Cells were either used directly for analysis or stored at −80 °C in heat-inactivated fetal bovine serum (FCS; Corning, 35-079-CV) with 10% v/v dimethylsulfoxide before analysis. Serum samples were stored at −80 °C before analysis.
Cytokine measurements
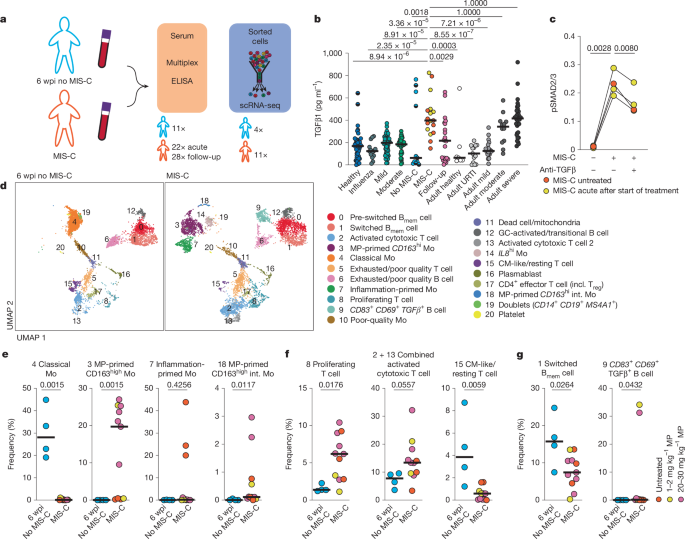
A bead-based multiplex cytokine array (Cytokine/Chemokine/Growth Factor 45-Plex Human ProcartaPlex Panel1, ThermoFisher Scientific) was used according to the manufacturer’s protocol. For TGFβ1 measurements, TGFβ1 was transformed to the bioactive form using 1 N HCl followed by neutralization with 1.2 N NaOH. A Human TGFβ1 Simplex ProcartaPlex kit (ThermoFisher Scientific) was used as previously described4. To normalize data between different runs, 4 to 16 samples were measured repeatedly per run. The average fold change difference was used to normalize data. The assay-specific lower limits of quantification are indicated in the graphs and listed in Supplementary Data 16.
Simple size-based western blot assay
T cells were isolated from PBMC using the human Pan T Cell Isolation Kit (Miltenyi Biotec) and 1–2 × 106 cells were seeded per well in TexMACS Medium (Miltenyi Biotec). pLenti-CMV-Blast-DNTGFBR2-HA was a gift from G.-P. Dotto (Addgene plasmid #130888; http://n2t.net/addgene:130888; RRID:Addgene_130888)63. Plasmid (1.6 µg) was overexpressed in 2 × 105 HEK293T cells (not mycoplamsa tested; originally purchased from ATCC, not reauthenticated) using Lipofectamine (Invitrogen) (DNA:lipofectamine ratio 1:3). As a control a non-expressing vector with a blasticidin selection marker was used. Six hours after transfection, cells were incubated with 10 µg ml−1 blasticitidin (ThermoFisher) for 18–48 h for positive selection of transfected cells. T cells and transfected HEK293T cells were serum-starved overnight prior to incubation with 10% v/v serum from patients with MIS-C for 30 min. For T cells as a control, patients’ sera were pre-incubated with antibodies directed against TGFβ1, TGFβ2 and TGFβ3 (50 µg ml−1, R&D Systems, MAB1835-SP) for 10 min. Cells were collected after 30 min and lysed using a buffer containing 0.5% v/v NP40 (Sigma), 150 mM NaCl, 50 mM TrisHCl, cOmplete protease inhibitors (Roche) and phosSTOP phosphatase inhibitors (Roche). Protein concentration was quantified by BCA (ThermoScientific) and 3 µg were loaded on a SimpleWestern 12–230 kDa separation plate (Bio-Techne) according to manufacturer’s protocol. For detection of SMAD2 or SMAD3 and phosphorylated SMAD2 or SMAD3 following antibodies were used: SMAD2/3 (D7G7) XP rabbit monoclonal antibody 8685 and phospho-SMAD2 (Ser465/467)/SMAD3 (Ser423/425) (D27F4) rabbit monoclonal antibody 8828 (both 1:100; both Cell Signaling). As a loading control rabbit anti-β-Tubulin (NB600-936; 1:100; Novus Biologicals) was used. Anti-Rabbit Secondary HRP Antibody (042-206; Bio-Techne; provided as working stock dilution from manufacturer) was used as a secondary antibody. The samples were acquired on a Wes instrument (Protein Simple; Bio-Techne) and the data were analysed using the Compass for SW software (Bio-Techne; Protein Simple version 3.1.7). Intensity of the signal was used to quantify relative abundance of phosphorylated SMAD2/3 and normalized to total SMAD2/3 signal.
Cell isolation from PBMC for single-cell sequencing
Frozen PBMC from 11 patients with MIS-C and 4 samples from patients 6 weeks after SARS-CoV-2 who did not develop MIS-C were thawed in RPMI 1640 (Gibco 61870‐044) with 20% v/v FCS (FCS; Corning, 35-079-CV). Cells were washed once in PBS (Th. Geyer) containing 1% w/v BSA (PAN Biotech P06-1391500), 2 mM EDTA (Invitrogen) and 2 µg ml−1 Actinomycin D (Sigma-Aldrich). Cells were incubated with human FcR-blocking reagent (1:50; Miltenyi Biotec) and stained using 1 of 4 TotalSeq anti-human Hashtags (C0251 C0252, C0253 or C0254) (1:250, clone LNH-94 + 2M2, BioLegend), CD3 FITC (1:100, clone UCHT1, in house), CD14 PerCP-Cy5.5 (1:50, clone QA18A22, BioLgend), CD38 APC (1:25, clone HIT2, Biolegend), HLA-DR APC-Vio770 (1:100, clone AC122, Miltenyi Biotec), CD19 V500 (1:200, clone HIB19, BD Biosciences) and CD27 PE (1:100, clone M-T271, BioLegend) at 4 °C for 30 min. Directly prior to sorting on a BD AriaII (BD Biosciences), DAPI (Sigma-Aldrich, 0.1 µg ml−1) was added.
Activated T cells were identified as DAPI−CD14−CD19−CD3+CD38+HLA-DRhi, memory B cells were identified as DAPI−CD14−CD3−CD19+CD27+ (and CD38hi for plasmablasts), and monocytes were identified as DAPI−CD14+ (Extended Data Fig. 2b).
For identifying methylprednisolone effects on activated cells and for activated cells from influenza infected patients, cells were sorted on a MA900 Multi-Application Cell Sorter (Sony Biotechnology). Instead of plasmablasts, lineage−HLA-DRhi cells were sorted (identified as DAPI−CD14−CD19−CD3−HLA-DRhi) (Extended Data Figs. 3b and 5a). For sorting of B cells and plasmablasts and analysing TCRVβ21.3 frequencies on T cells, frozen cells were prepared for sorting as mentioned above and stained with CD3 FITC (1:100, clone UCHT1, in house), CD27 PE (1:100, clone M-T271, BioLegend), HLA-DR PerCP-Cy5.5 (1:50, clone L243, BioLegend), CD21 PE-Vio770 (1:75, clone HB5, Miltenyi Biotec), CD14 VioBlue (1:200, clone TÜK4, Miltenyi Biotec) or CD14 AlexaFluor700 (1:500, clone TM1, in house), CD19 BV510 (1:25, clone HIB19, BioLegend), CD38 APC (1:25, clone HIT2, Biolegend), TCRVβ21.3 APC-Vio770 (1:100, clone REA894, Miltenyi Biotec). Directly prior to sorting DAPI (Sigma-Aldrich, 0.1 µg ml−1) was added. Cells were sorted on a MA900 Multi-Application Cell Sorter (Sony Biotechnology) or a Cytek Aurora (Cytek Biosciences). B cells were identified as DAPI−CD14−CD3−CD19+CD21+ and plasmablasts as DAPI−CD14−CD3−CD19+CD27+CD38hi (Extended Data Fig. 9a). T cells were identified as DAPI−CD14−CD19−CD3+, and activated T cells were identified as DAPI−CD14−CD19−CD3+CD38+HLA-DRhi (Extended Data Fig. 7l,m), Sorted cells were counted using a MACSQuant Analyzer 16 (Miltenyi Biotec) and processed for scRNA-seq using 10X Genomics technology.
Single-cell RNA library preparation and sequencing
Single-cell RNA library construction and sequencing was done as previously described4,30. In brief, Chromium Next GEM Single Cell 5′ reagent kits v2 (dual index) with feature barcode technology for cell surface protein (CITE) mapping (10X Genomics) were used according to the manufacturer’s protocol. Final CITE-seq libraries were generated after index PCR with dual Index Kit TN Set A (10X Genomics) and final GEX, TCR and B cell receptor (BCR) libraries were generated after fragmentation, adapter ligation and final index PCR with a dual Index Kit TT Set A (10X Genomics). Libraries were quantified using a Quibit HS DNA assay kit (Life Technologies) and fragment sizes were determined using a HS NGS Fragment (1–6,000 bp) kit (Agilent).
Sequencing was performed on a NextSeq2000 sequencer (Illumina) using a P3 reagent cartridge (100 cycles) (Illumina) with the following recommended sequencing conditions: read 1: 26 nt, index 1: 10 nt, read 2: 90 nt, index 2: 10 nt.
Single-cell transcriptome analysis
Raw sequence reads were processed using cellranger (version 5.0.0), including the default detection of intact cells based on the EmptyDrops method64. Demultiplexing, mapping, detection of intact cells, as well as quantification of gene expression was performed using cellranger’s count pipeline in default parameter settings with refdata-cellranger-hg19-1.2.0 as reference, Hashtag 1–4 as feature reference and expected number of 3,000 cells per sample. Of note, the used reference does not contain immunoglobulin genes and TCR genes as defined by the respective biotype, except for TRBV11-2 which was removed in addition. This led to 8,531, 4,356 and 7,644 intact cells for MIS-C pools and 8,060 for controls. Next, cellranger’s aggr was used to merge the libraries without size normalization and further analysed in R (version 4.1.2) using the Seurat package (version 4.0.5)65. In particular, the transcriptome profiles were read using Read10x and CreateSeuratObject and log-normalized using NormalizeData. A UMAP was computed using ScaleData, RunPCA to compute 50 principal components and RunUMAP using 1:50 dimensions. Transcriptionally similar clusters were identified by shared nearest neighbour modularity optimization using FindNeighbors with pca as reduction and 1:50 dimensions as well as FindClusters with resolutions ranging from 0.1 to 1.0 in 0.1 increments using FindCluster. Clustering with a resolution of 0.6 (Extended Data Fig. 2c) was judged to best reflect the transcriptional community structure, by visually inspecting the percentage of mitochondrial genes, UMI counts, number of identified genes, housekeeping genes, TCRs and BCRs, as well as expression of typical marker genes projected on the UMAP (Extended Data Fig. 2d–i). Clusters were annotated based on the expression of core marker genes (Extended Data Fig. 2f–i), top differentially expressed genes (Supplementary Data 1, 2 and 17), cell cycle scores, derived from the scaled expression of genes associated with different cell cycle stages using the Loupe Browser (version 5 and 6, 10X Genomics). To further refine cell identification, the identification of isotypes for B cells by BCR sequencing was used to distinguish between pre-switched and switched memory B cells (Extended Data Fig. 2i). TCR sequencing was additionally used to identify T cells. Clusters comprising low quality cells as well as clusters comprising contaminations were not considered in further analyses. Samples were demultiplexed using hashtag reads after log-normalization and manual distinction of positively tagged cells using histograms.
Single-cell immune profiling
TCR annotations were based on single-cell immune profiling. In particular, raw sequence reads were processed by cellranger (version 5.0.0) with vdj in default parameter settings for demultiplexing and assembly of the TCR sequences using refdata-cellranger-vdj-GRCh38-alts-ensembl-2.0.0 as reference. Annotations of the TCR were assigned to the corresponding cells in the single-cell transcriptome analysis by identical cellular barcodes. In case of multiple contigs, the most abundant, productive and fully sequenced contig for the α and β or γ and δ chain was considered, respectively. Noteworthy, cellranger mislabelled TRBV11-2 with TRBV11-1 annotations as judged by multiple alignment of the sequences of the respective contigs to the references of TRBV11-1 and TRBV11-2 as well as mapping of transcriptome to the TRBV11-2 gene locus.
Gene set enrichment analysis
GSEA was performed as previously described57 for each individual cell based on the difference to the mean of log-normalized expression values of monocytes, B cells including plasmablasts, or T cells manually selected using cloupe (version 6.3.0) in the analysed set as pre-ranked list and 1,000 randomizations66,67. The GSEA was performed separately for T cells, B cells (with plasmablasts) and monocytes. Permutations are conducted by gene set to calculate NES, FDRs, values and core genes for each cell. The number of significant cells for each set and core genes are represented in Supplementary Data 3. Significant up- or downregulation was defined by a FDR ≤ 0.50 or with an FDR ≤ 0.25 (the recommended FDR for bulk sequencing)66 as an additional control (Extended Data Fig. 6) and by a normalized P value < 0.05. For visualization, NES for significant cells were plotted. The GSEA was performed for indicated cells using Hallmark gene sets, our previously published TGFβ NK-cell gene set4 and previously defined immune transcription modules59. Hallmark gene sets were obtained from the MSigDB Collections68. The GSEA results were visualized by projection of the NES scores on UMAPs as well as on violin plots of the NES score of significant enriched cells (positive NES score).
Ex vivo stimulation of isolated mononuclear cells
Antigen-specific restimulation experiments were performed in RPMI 1640 medium (Gibco), supplemented with 5% v/v human AB serum (Sigma-Aldrich) or 10% v/v patients’ sera, 100 U ml−1 Penicillin/Streptomycin (Gibco) and 2 mM l-glutamine (HyClone). For antigen-reactivity measurements, a total of 5 × 105 PBMCs were stimulated for 16 h with the following antigens. For SARS-CoV-2: PepTivator SARS-CoV-2 Prot_N, PepTivator SARS-CoV-2 Prot_S, PepTivator SARS-CoV-2 Prot_M, Omicron-specific pepTivator SARS-CoV-2 Prot_S B.1.1.529 (all 60 nM per peptide; Miltenyi Biotech). For EBV: PepTivator EBV Consensus, premium grade (60 nmol per peptide; Miltenyi Biotech). For HSV-1: PepTivator HHV1 Envelope Glycoprotein D, research grade (60 nmol per peptide; Miltenyi biotech). For HSV2: PepMix HSV2 (gD) (15 nM per peptide; JPT peptide technology). For HHV-6 (U54) Peptide Pool (15 nM per peptide; peptides&elephants). For AdV: PepTivator AdV Select (60 nM per peptide; Miltenyi Biotech). Other peptides used were Staphylococcus enterotoxin B (SEB; Sigma-Aldrich) 1 µg ml−1 or peptide pools of cytomegalovirus (CMV) pp65 (60 nM per peptide; Miltenyi Biotech). Stimulation was performed in the presence of 1 μg ml−1 CD40 and 1 μg ml−1 CD28 functional grade pure antibody. When used, patient sera were pre-incubated with antibodies directed against TGFβ1, TGFβ2 and TGFβ3 (50 µg ml−1, R&D Systems, MAB1835-SP) for 10 min. For EBNA2-peptide-specific T cells the EBNA2275–295 peptide PRSPTVFYNIPPMPLPPSQL (10 µg ml−1) or the EBNA2279–290 peptide TVFYNIPPMPL (5 µg ml−1) were custom-made (peptides&elephants, purity ≥80%) and at least 2 × 107 PBMCs were stimulated.
Cells were incubated with FcR-blocking reagent (1:50, Miltenyi Biotec) and stained for flow cytometric analysis with CD14 VioBlue (1:200, clone REA599, Milenyti Biotec), CD19 VioBlue (1:100, clone REA675, Miltenyi Biotec), CD45RO BV510 (1:50, clone UCHL1, BD Biosciences), CD3 PE Cy5 (1:200, clone UCHT1, BioLegend), CD8a BV650 (1:100, clone RPA-T8, BioLegend), CD4 PerCPeFluor710 (1:200, clone SK3, eBioscience), CD69 APC-Cy7 (1:100, clone FN50, BioLegend), CD154 PE (1:100, clone REA238, Miltenyi Biotec) CD137 PE Cy7 (1:100, clone 4B4-1, BioLegend) as mentioned above. Directly prior to analysing on a MACSQuant Analyzer 16 (Miltenyi Biotec) using MACSQuantify software, DAPI (Sigma-Aldrich, 0.1 µg ml−1) was added. CD69+CD4+ memory T cells were defined as DAPI−CD14−CD19−CD3+CD45RO+CD8−CD4+CD69+. CD69+CD8+ memory T cells were defined as DAPI−CD14−CD19−CD3+CD45RO+CD4−CD8+ and CD69+. CD154+CD69+CD4+ memory T cells were defined as DAPI−CD14−CD19−CD3+CD45RO+CD8−CD4+CD154+ and CD69+CD154+CD69+CD8+ memory T cells were defined as DAPI−CD14−CD19−CD3+CD45RO+CD4−CD8+CD154+CD69+. CD137+CD69+CD8+ memory T cells were defined as DAPI−CD14−CD19−CD3+CD45RO+CD4−CD8+CD137+CD69+ (Extended Data Fig. 7a,c). For EBNA2275–295– and EBNA2279–290-specific T cells, cells were stained with CD14 VioBlue (1:200, clone REA599, Milenyti Biotec), CD19 VioBlue (1:100, clone REA675, Miltenyi Biotec), CD45RO BV510 (1:50, clone UCHL1, BD Biosciences), CD3 PE Cy5 (1:200, clone UCHT1, BioLegend), CD8a BV650 (1:100, clone RPA-T8, BioLegend), CD4 PerCPeFluor710 (1:200, clone SK3, eBioscience), CD69 APC (1:50, clone FN50, Miltenyi Biotec), CD154 PE (1:100, clone REA238, Miltenyi Biotec) and TCRVβ21.3 APC-Vio770 (1:100, clone REA894, Milteyi Biotec). CD4+ or CD8+ memory T cells and CD154+ CD69+ CD4+ or CD8+memory T cells were analysed for expression of TCRVβ21.3 (Extended Data Fig. 8c) using FlowJo software 10.8.1 (TreeStar).
HLA haplotyping
DNA was extracted from whole blood or buccal swabs (eSwab, Copan) using the QIAamp DNA Micro kit (QIAGEN), from patients with MIS-C and heathy controls (children without MIS-C and adults used for the generation of virus-specific T cell libraries). The DNA was subjected to HLA genotyping for HLA-A, HLA-B, HLA-C, DRB1, DRB3, DRB4, DRB5, DQA1, DQB1 and DPB1. HLA typing was performed using the Protrans NGS for 7 Loci kit (Protrans Medical Diagnostics) by next generation sequencing. The isolated genomic DNA was used in eight multiplex PCR reactions and labelled with Nextera XT index primer kits (Illumina). After group-specific pooling, followed by CleanPCR Beads purification (Beckman Coulter) and normalization (Quant-iT PicoGreen, Thermo Fischer Scientific), a MiSEQ library was prepared and sequenced on an Illumina MiSeq platform. HiType software (Inno-Train) based on the most recent HLA database was used for data analysis.
TCR library preparation of virus-specific T cells by ARTE assay
At least 1 × 107 total PBMCs were stimulated as mentioned above. After stimulation, cells were stained with TotalSeq anti-human Hashtags as previously mentioned, followed by CD154 MACS enrichment according to the manufacturer’s protocol (CD154 MicroBead Kit, human; Miltenyi Biotec). After MACS, cells were incubated with human FcR-blocking reagent (1:50, Miltenyi Biotec) and stained for FACS enrichment with PE-anti Biotin (1:100, clone Bio3-18E7, Miltenyi Biotec), CD154 PE (1:40, clone REA238, Miltenyi Biotec), CD14 VioBlue (1:200, clone REA599, Milenyti Biotec), CD19 VioBlue (1:100, clone REA675, Miltenyi Biotec), CD3 PE Cy5 (1:200, clone UCHT1, BioLegend) and CD69 APC (1:100, clone REA824, Miltenyi Biotec). DAPI (Sigma-Aldrich, 0.1 µg ml−1) was added and cells were sorted on a MA900 Multi-Application Cell Sorter (Sony Biotechnology) according to scatter properties, viability (DAPI−) and dump− (CD14 and CD19) specifically for CD3+CD154+CD69+ antigen-specific cells (Extended Data Fig. 8a). A heat map was generated using the R package pheatmap.
EBV killing assay
EBV-immortalized B cells (LCL) from healthy donors were generated as previously described69. In brief 5–10 × 106 PBMCs were seeded in RPMI 1640 medium (Gibco), supplemented with 10% v/v FCS (Corning or Biowest) and 250 µl EBV supernatant 4 μg ml−1 CpG and 0.5 μg ml−1 Cyclosporin A and passaged subsequently. From the same donors, T cells were isolated from PBMC using the Human Pan T cell Isolation Kit (Miltenyi Biotec), and incubated with human FcR-blocking reagent (1:50, Miltenyi Biotec) and stained with CD3 FITC (1:100, clone UCHT1, in-house), CD8a BV650 (1:100, clone RPA-T8, BioLegend), CD4 PerCPeFluor710 (1:200, clone SK3, eBioscience), TCRVβ21.3 APC-Vio770 (1:100, clone REA894, Miltenyi Biotec). Prior to sorting, DAPI (Sigma-Aldrich, 0.1 µg ml−1) was added. Cells were sorted on a MA900 Multi-Application Cell Sorter (Sony Biotechnology). Viable (DAPI−) and VioBlue− CD3+ cells were further sorted according to CD4, CD8 and TCRVβ21.3 expression (Extended Data Fig. 8d). Forty thousand cells were seeded per well and polyclonally expanded using Dynabeads human T-Activator CD3/CD28 (ThermoFisher) at a bead-to-cell ratio of 1:1 to generate enough material for the killing assays. For CD4 cells, 50 U ml−1 human recombinant IL-2 (Peprotech) was added and for CD8 expansion, 10 ng ml−1 IL-15 (Peprotech) was added to the medium (TexMACS Medium (Miltenyi Biotec) with 5% human AB serum (Sigma-Aldrich) and 100 U ml−1 Penicillin/Streptomycin (Gibco)). LCLs were marked with CFSE (1:2000, BioLegend). After expansion, viable, CFSE-marked LCLs and expanded T cells from each donor were counted after staining with DAPI and 1 × 104 viable LCLs were seeded with respective numbers of viable T cells from the same donor and centrifuged for 2 min at 200 rpm. LCLs were co-cultured for 4 h with CD8+ T cells and 24 h with CD4+ T cells at 37 °C 5% CO2. To detect recent degranulation, a CD107a assay was used. For this assay, CD107a AlexaFluor647 (1:3000, clone H4A3, BioLegend) and Brefeldin A (5 µg ml−1, BioLegend) were added for the last 4 h, as CD107a is only briefly expressed on the surface during degranulation44. Cells were stained with fixable viability dye ZombieAqua (1:400, BioLegend) or Viobility 405/452 Fixable Dye (1:100, Miltenyi Biotec), CD3 PE Cy5 (1:200, clone UCHT1, BioLegend), CD8a BV650 (1:100, clone RPA-T8, BioLegend), CD4 PerCPeFluor710 (1:200, clone SK3, eBioscience) and TCRVβ21.3 APC-Vio770 (1:100, clone REA894, Miltenyi Biotec). Afterwards, cells were fixated and permeabilized with the True Nuclear staining kit (BioLegend) and stained with active caspase-3 AlexaFluor647 (1:50, BD Bioscience). Viable LCLs were defined as CFSE+, fixable viability dye− and active caspase-3− cells (Extended Data Fig. 8e). Cell counts were normalized to LCLs cultured without T cells and CD107a (Extended Data Fig. 8f) staining was normalized to T cells cultured without LCL to subtract background expression of CD107a.
Antibody quantification
Serum IgG-antibody levels against HSV-1 (Abnova, KA0229), HSV2 (Abnova, KA0231), EBNA1 (abcam, ab108731), CMV (Abnova, KA1452), HHV-6 (Abnova, KA1457) and AdV (Creative Diagnostics, DEIA2382) and serum IgM antibody levels against HSV-1/2 (Abnova, KA4842), EBNA1 (Abnova, KA1449), CMV (Abnova, KA0228), HHV-6 (Creative Diagnostics, DEIABL57) and AdV (Creative Diagnostics, DEIA1767) were measured in first serum samples obtained from patients according to manufacturer’s manuals. A provided working stock solution of the antibodies was used without further dilution. Arbitrary units were calculated based on reference controls. For age-matched comparison of seroprevalences, patients were grouped by age according to published age ranges50,70,71,72. Expanded humoral profiling data was obtained from a previously published MIS-C cohort49. Published cohorts or control cohorts, including healthy children and children with SARS-CoV-2 infection who did not get MIS-C, were adjusted to match the age distribution of the test population (Supplementary Data 12). In hospital data for virus serology was determined by accredited Immunoblotting Assays for EBV-IgM and –IgG antibodies and enzyme immunoassays by automatic analysis (Liaison, Diasorin and Architect, Abbott).
EBV transcript detection in single-cell dataset
For detection of EBV reactivation, unmapped reads from the transcriptome analysis were extracted from the .bam files from the transcriptome analysis and mapped to the human gamma herpesvirus 4 references (NCBI_Assemblies:GCF_002402265.1, GCF_000872045.1.). Specific binding as well as correct gene annotation was verified using blast with standard databases. EBV UMI counts were assigned to the corresponding cells in the single-cell transcriptome analysis by identical cellular barcodes.
EBV load in cell-free plasma
EBV viral DNA (elution volume 50 µl) was extracted from 200 µl of plasma sample using an EasyMag extractor (bioMérieux, Marcy-l’Etoile, France) following the manufacturer’s instructions. The presence and viral load of EBV were then determined using the EBV R-GENE kit (available for research use only, not for diagnostic, 69-002B; bioMérieux). Log of EBV DNA copy number per ml (log copies ml−1) plasma are used to describe EBV viral load. The reported LOD was 1.6 log copies ml−1.
EBV reactivation assay
For EBV reactivation, LCLs were incubated in RPMI 1640 (Gibco 61870‐044) with 10% v/v FCS (Biowest S1600-500) or 10% v/v patient’s serum and 1% v/v Penicillin/Streptomycin (Gibco 15140-122). Recombinant human TGFβ1 (10 ng ml−1; PeproTech, 100-21) was used as a positive control. If TGFβ was neutralized, the patient’s serum was pre-incubated for 10 min with 50 µg ml−1 of antibodies directed against TGFβ1, TGFβ2 and TGFβ3 (R&D Systems, MAB1835-SP). Cells were collected after 24 h and total RNA was extracted using the RNeasy Plus Micro Kit (Qiagen). cDNA was transcribed using the TaqMan reverse transcription kit using random hexamers (Life Technologies). TaqMan PCR was performed using human HPRT1 primers and probes of TaqMan gene expression assays (Life Technologies) as well as the following combination of primers and probes for the transcription factor BZLF1, which induces the lytic EBV replication cycle, forward: 5′-CTCAACCTGGAGACAATTCTACTGT-3′, reverse: 5′-TGCTAGCTGTTGTCCTTGGTTAG-3′, probe: 5′-FAM-CTGCTGCTGCTGTTTG-3′NFQ (Life Technologies). Samples were measured on a QuantStudio 5 Real-Time PCR System (Applied Biosystems).
Statistical analysis and reproducibility
GraphPad Prism (v8.00 to v9.4.1) for Windows (GraphPad Software) was used for statistical analysis of the data. If applicable, all data were plotted as individual values and the median was given as data summary. For two-group comparisons of non-normal distributed data, a two-tailed Mann–Whitney U-test was used. If normal distribution could be assumed a two-tailed t-test was used. For paired samples, the paired-test equivalence was used. For multiple group comparisons, two-tailed non-parametric ANOVA (Kruskal–Wallis test) was used, followed by a Dunn’s multiple comparison test with correction for multiple comparisons. Correlations were calculated using a Spearman correlation. Testing for significance in GSEA: if only positive enrichment was quantified, a two-tailed Mann–Whitney U-test was used; otherwise, to test for positive and negative enrichment in bimodal data a two-tailed Fisher’s exact test was used. Testing for increased seroprevalences of latent virus infections: a one-tailed Fisher’s exact test was used. The significance threshold for all tests was set to 0.05 and P values are provided with up to 4 significant figures where applicable.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.